
邮箱:zjucps@zju.edu.cn
地址:浙江省杭州市西湖区余杭塘路866号,浙江大学紫金港校区
Copyright © 2018 浙江大学药学院版权所有
技术支持:YONCC

先导化合物的发现与优化在新药研发过程中至关重要,高质量的先导化合物能够大大缩短药物探索的时间,提高成药的可能性。全新药物设计可以通过不同的生成算法对类药空间进行深入探索和发掘,从而生成出大量化学结构有效且新颖的分子,但如何能够在避免无效和重复采样的前提下提高采样分子对靶标蛋白的亲和性是当前研究难点。
2022年6月,浙江大学药学院&智能创新药物研究院侯廷军团队、中南大学曹东升团队、华东理工大学李洪林团队联合在《药物化学杂志》(Journal of Medicinal Chemistry)发表论文“RELATION: A Deep Generative Model for Structure-based De Novo Drug Design”,提出了一种能够在分子生成过程中考虑到蛋白-配体相互作用的深度学习全新药物设计新方法。

为解决目前全新药物设计方法采样的分子无法同时兼顾化学结构的质量和对蛋白亲和力,作者同时使用百万量级的分子库以及蛋白配体集合数据对变分自编码器进行训练,在引入双向迁移学习之后,隐藏层的采样能够同时兼顾生成分子的骨架片段的新颖性以及对靶标蛋白的亲和性。作者在方法中还提供了药效团约束生成以及贝叶斯优化采样等模块,可定制化生成药效团匹配度更高以及对靶标亲和力表现更优的分子。
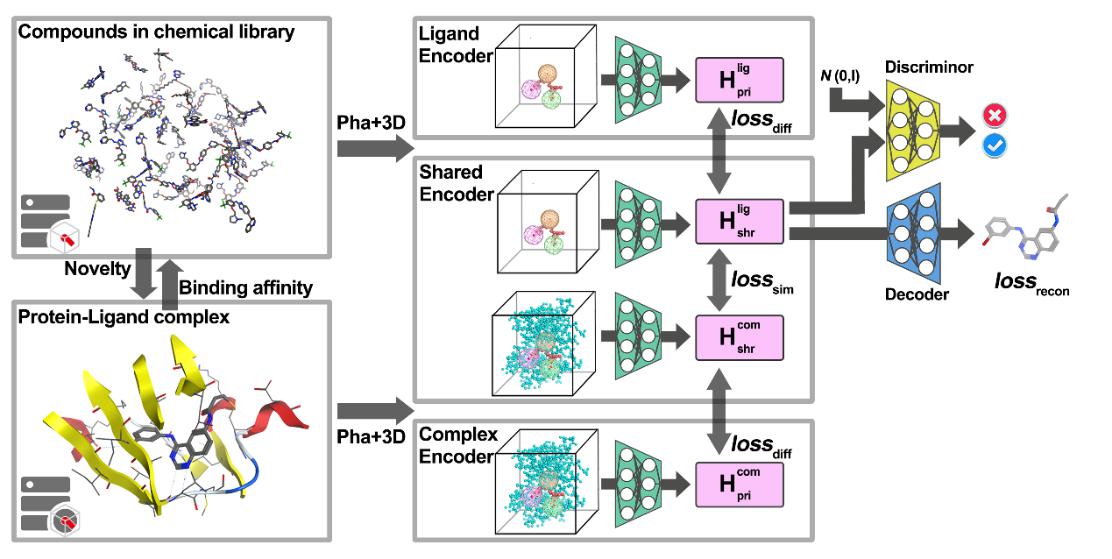
图1 RELATION方法的工作流程图
在AKT1与CDK2的抑制剂全新设计的任务测试中,RELATION模型既能生成有效、新颖且多样的分子,并且能够保证生成的分子对靶标具有一定的亲和力;随着基于对接打分的BO采样以及药效团约束模块用于RELATION模型,RELATION模型能够使得生成的分子同时具有更好的药效团匹配度和对接表现。结果表明RELATION模型是一种极具竞争力的深度学习全新药物设计模型,有望为先导化合物的设计和发现提供全新的策略。
浙江大学药学院&智能创新药物研究院为本论文的第一署名单位,浙江大学药学院博士生王明阳和腾讯量子实验室谢昌谕博士为共同第一作者,浙江大学侯廷军教授、中南大学曹东升教授、华东理工大学李洪林教授为共同通讯作者。
原文链接:https://doi.org/10.1021/acs.jmedchem.2c00732